


Abstrakt
25 yaşında, əvvəllər heç bir şikayəti olmayan gənc oğlan son 2 ayda 3 dəfə baş verən qısa müddəli bayılmalardan şikayət edir. Sözlərinə görə bayılmadan əvvəl bədənində titrətmə hiss edir. EKQ-də mədəcik ekstrasistolları ilə yanaşı sağ döş aparmalarında neqativ T dişcikləri qeydə alındı. Ürəkdə hər hansı struktur dəyişlikliyin olub olmadığını bilmək üçün 2D, M, TDI-PW rejimlərindən istifadə etməklə exokardioqrafik müayinə aparıldı. Müayinə zamanı sağ mədəciyin (SağM) sərbəst divarında çapıq toxumasına bənzər hiperexogen sahə, sağ mədəciyin genişlənməsi və sistolik funksiyasının zəifləməsi aşkarlandı. EKQ-nin 24 saatlıq Holter monitorlamasında çoxlu miqdarda (politop, qrupşəkilli, triplet) mədəcik ektopiyası qeydə alındı. Bu nəticələrə asasən xəstədə sağ mədəciyin aritmogen displaziyası (ARVD, ARVC) olmasına şübhə yarandı və ürəyin MRT olunmasına qərar verildi.
Əsas mətn
Klinik müşahidə
25 yaşında, əvvəllər heç bir şikayəti olmayan gənc oğlan son 2 ayda 3 dəfə baş verən qısa müddəli bayılmalardan şikayət edir. Sözlərinə görə bayılmadan əvvəl bədənində titrətmə hiss edir. Aparılan müayinə ürəyin auskultasiyasında və arterial təzyiqdə hər hansı bir anormallıq aşkarlamadı. Nəbz ardıcıl baş verən ekstrasistollar səbəbindən aritmik kimi dəyərləndirildi. İlkin olaraq xəstənin 12 aparmalı standart sakitlik EKQ-si çəkildi (şəkil 1). EKQ-də mədəcik ekstrasistolları ilə yanaşı sağ döş aparmalarında neqativ T dişcikləri qeydə alındı.
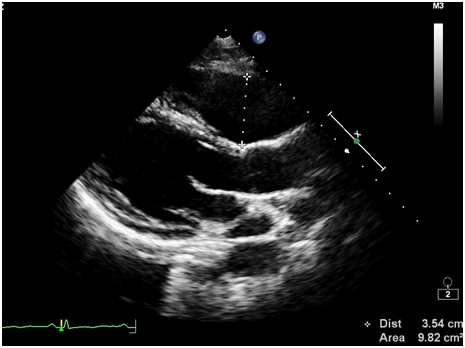
Ürəkdə hər hansı struktur dəyişlikliyin olub olmadığını bilmək üçün 2D, M, TDI-PW rejimlərindən istifadə etməklə exokardioqrafik müayinə aparıldı. Müayinə zamanı sağ mədəciyin (RV) sərbəst divarında çapıq toxumasına bənzər hiperexogen sahə, sağ mədəciyin genəlməsi və sistolik funksiyasının zəifləməsi (FAC – 30.5%, N > 35%) aşkarlandı (şəkillər 2-8). Bunun ardınca təyin olunan EKQ-nin 24 saatlıq Holter monitorlamasında çoxlu miqdarda (politop, qrupşəkilli, triplet) mədəcik ektopiyası qeydə alındı (şəkil 9). Bu nəticələrə asasən xəstədə aritmogen sağ mədəcik displaziyasına (ARVD, ARVC) şübhə yarandı və ürəyin MRT olunmasına qərar verildi. Ürəyin MRT müayinəsində aritmogen displaziyaya uyğun görüntülər qeydə alındı.
Xəstəyə rəhbər tövsiyələrə uyğun olaraq elektrofizioloji müayinə və sonradan kardioverter-defibrilyator taxılması məsləhət görülmüşdür. Lakin o, elektrofizioloji müayinədən imtina etmişdir. Hal-hazırda yalnız dərman müalicəsi qəbul edir: sotalol, gündə 1 dəfə. 6 ay sonra Holter müayinəsi aparılmışdır. Ağır dərəcəli aritmiya qeydə alınmamışdır.
Müzakirə
Ümumi məlumat. Aritmogen sağ mədəcik displaziyası/kardiomiopatiya (ARVD/ARVC) anadangəlmə kardiomiopatiya olub ona struktur və funksional SağM anormallıqları xasdır ki, bu da mədəcik aritmiyaları ilə nəticələnir.[1] Bu, gənc yaşlarda qəfləti ürək ölümünün mühüm səbəblərindəndir. Rastgəlməsi gənclərdə baş verən bütün hallar üçün 11%, gənc atletlərdə baş verən hallar arasında isə 22% təşkil edir. [2, 3]
ARVD ilk dəfə 1977-ci ildə təsvir edilmişdir və ÜST-nin 1995-ci ildə kardiomiopatiyalara dair verdiyi təsnifata daxil edilmişdir. [4] O vaxtdan bu xəstəliyin etiopatogenezinin, diaqnozunun və müalicəsinin anlaşılmasında mühüm irəliləyişlər olmuşdur. [5]
Patofiziologiyası. ARVD olanlarda struktur anormallıqlar SağM miokardının yağ infiltrasiyası və fibrozla əvəz olunmasıdır. Bu, SağM dilatasiyası və disfunksiyasına səbəb olur. Sol mədəcik (SolM) prosesə daha az cəlb olunur, mədəcikarası çəpər isə nisbətən daha az təsirə məruz qalır. [6] Lakin 200 proband üzərindəki kohortda Sen-Chowdhry və h.m. müəyyən etmişlər ki, əhəmiyyətli SağM disfunksiyası həm də prosesə SolM cəlbi ilə də müşayiət oluna bilər. [7] SolM prosesə cəlb olunduqda proqnoz ağırlaşır. [8]
Miokard toxumasının itirilməsi mexanizmlərinə aşağıdakılar daxildir:
Apoptoz (proqramlı hüceyrə ölümü)
Iltihab, artmış fibroz və funksiyanın itirilməsi
Miokardın yağ toxuması ilə əvəz olunması
Etiologiyası. ARVD anadangəlmə pozulma kimi artıq bətndaxili inkişafda baş verir. Ailəvi hallar bütün halların 30-90%-ini təşkil edir. [9] Digər hallarda bu virus infeksiyası (miokardit) kimi qazanılma etiologiyaya və ya naməlum irsiyyətə malik ola bilər. Nəzərə almaq vacibdir ki, genetik meyli olanlarda miokarditin inkişafı daha çox hallarda baş verə bilər.
Xəstəlik daha tez-tez fəal fərdlərdə baş verir ki, onlarda da əgər SağM genetik meyllidirsə mexanik yerdəyişmə stresi (shear stress) hüceyrə membranı zədələnməsinə, iltihaba və fibroza səbəb ola bilər.
Genetikası. ARVD bir çox hallarda ailəvi genetik pozulma olub coğrafi klasterlənməyə malikdir. İrsiyyətdə daha çox ailə üzvləri arasında 20-35% autosom dominantlıq üstünlük təşkil edir. [10, 11] İtaliyanın Veneto regionunda yaşayan insanlarda rastgəlmə yüksəkdir. ÜST-nin 1996-cı ildə kardiomiopatiyalara dair verdiyi təsnifata görə Naksos xəstəliyi aritmogen sağ mədəcik displaziyası/kardiomiopatiyaların resessiv (ARVD/ARVC) forması kimi nəzərdə tutulmuşdur (şəkil 13). Qeyd edək ki, Naksos adası Yunanıstana aiddir. Bu xəstəliyin əlamətlərinə palmoplantar keratoz və qoyun yunu kimi saçlar aid edilir.
Genetik mutasiya 17q21 xromosomunda baş verir və penetrantlıq (genin fenotipdə üzə çıxma göstəricisi) demək olar ki, 100% təşkil edir. [12, 13] ARVD-də genetik anomaliyalar 1, 2, 3, 6, 7, 10, 12 və 14-cü xromosomlarda yerləşir. Lakin burada yalnız bir unikal genetik anomaliya olmadığı üçün ARVD şübhəsi olan xəstələrin və ailələrin qiymətləndirilməsində çətinlik vardır. Məsul genlərə plakoqlobin (JUP), desmoplakin (DSP), plakofilin-2 (PKP2), desmoqlein-2 (DSG2), desmokollin-2 (DSC2) və digərləri aid edilir. [14, 15, 16]
2011-ci ildə Ürək Ritmi Cəmiyyəti və Avropa Ürək Ritmi Assosiasiyasının kardiomiopatiyaların genetik testləməsinə dair konsensus sənədi dərc edilmişdir.[17] Naksos xəstəliyinə dəri və ürək fenotipinin dəyişilməsi xasdır. Burada saçlar qoyun yunu formasında (A) olur, ətraflarda palmoplantar keratoderma (B və C) kimi prinsipial dəri anormallığı baş verir. Ürəkdə xəstəlik ARVD ilə öz əksini tapır. Miokardın itirilməsi və SağM miokardının fibroz və yağ toxuması ilə əvəzlənməsi (D) gecikmiş fəallığa (sakitlik EKQ-sində epsilon dalğaları) (E) səbəb olur ki, bu da reentrant mədəcik aritmiyalarına meyl törədir (F).
Epidemiologiyası. ARVD-nin dəqiq rastgəlmə göstəricisi diaqnostik problemlərə görə onun gizli formaları heç də həmişə aşkarlanmadığı üçün naməlum qalır. [18] Hesablanmışdır ki, bu xəstəliyə populyasiyada hər 1000-5000 nəfərdən 1-də və daha çox Yunanıstan və İtaliya mənşəyi olanlarda rast gəlinə bilər. [19]
ABŞ-dan olan 100 xəstədən ibarət kohortda orta yaş 26 il olmuşdur ki, bunlardan da 51%-ini kişilər, 49%-ini isə qadınlar təşkil etmişlər. Diaqnozun qoyulmasına keçən orta müddət xəstəliyin başlanğıcından sonra bir il olmuşdur.[20]
Klinik əlamətləri. ARVD asimptomatik olmaqdan başlayıb hər iki mədəciyin hipertrofiyası/yaxud qəfləti ürək ölümünədək ən müxtəlif və geniş spektrli əlamətlərlə özünü biruzə verə bilər.
Xəstəliyin aşağıdakı ümumi simptomları haqqında məlumat verilmişdir [20, 21]:
Ürəkdöyünmə (27%-67%)
Sinkop (26%-32%)
Qəfləti ürək ölümü (10%-26%)
Atipik döş qəfəsi ağrısı (27%)
Təngnəfəslik (11%)
Mədəcik aritmiyaları baş verməsi səbəbindən ürəkdöyünməyə daha tez-tez rast gəlinir. Xəstəliyin ağırlığından asılı olaraq yalnız izolə mədəcik ektopiyaları və ya davamsız/davamlı mədəcik taxikardiyaları, mədəcik fibrilyasiyası və qəfləti ölüm baş verə bilər.
Proqressiv SağM disfunksiyası təngnəfəslik və ayaqlarda şişkinliklə müşayiət oluna bilər. Bəzi ağır hallarda prosesə SolM də cəlb olunur ki, bu da hər iki mədəciyin hipertrofiyası ilə gedən ürək çatışmazlığı və dilatasion kardiomiopatiya təəssüratı oyadır. Supraventrikulyar aritmiyalara, atrial titrəmə və fibrilyasiya da daxil olmaqla, 25% halda rast gəlinir. [22]
Fiziki yük mədəcik aritmiyalarına və qəfləti ürək ölümünə səbəb ola bilər. ARVD Şimali İtaliyada gənc atletlər arasında bütün qəfləti ürək ölümlərinin 22%-ni təşkil edir.[2, 23] ABŞ-dabaş verən286 qəfləti ürək ölümü halının əsassəbəbihipertrofikkardiomiopatiyaolmuşdur, ARVDbuhallarınyalnız 4%-initəşkiletmişdir.
Differensial diaqnostikası.
Idiopatik sağ mədəcik taxikardiyası. Fiziki yük təsirindən sol ayaqcıq bloku morfologiyalı mədəcik taxikardiyası baş verməsi səbəbindən ARVD ilə oxşarlığa malik olsa da xoşxassəli gedişə malikdir. Burada ürək strukturu normal olur, ailə anamnezi yoxdur və qəfləti ürək ölümü baş vermir. SağM taxikardiyasında EKQ-də V2 -V5 aparmalarında T dişcikləri yuxarı yönəlir və epsilon dalğaları olmur. [24, 25]
Uhl anomaliyası. Nadir hallarda baş verən pozulma olub SağM divarının nazilməsi və əzələ toxumasının hissəvi yaxud tam itirilməsi xasdır (perqament SağM).[26] Miokard müxtəlif dərəcəli fibroza məruz qalır.
Dilatasion kardiomiopatiya.Hər iki mədəciyin dilatasiyası və durğunluq ürək çatışmazlığı SolM cəlbi ilə gedən ARVD kimi şərh oluna bilər. ARVD üçün xarakterik EKQ və ürək MRT-si anormallıqları iki xəstəliyi bir-birindən ayırd etməyə kömək edir.
Laboratoriya tədqiqatları. ARVD müxtəlif cür üzə çıxdığı və risk altında olan populyasiyalara böyük diqqət tələb etdiyi üçün ciddi problem törədir. Əvvəllər diaqnoz ölümdən sonrakı tapıntılara və ya endomiokardial biopsiyada histoloji təsdiqə əsaslanırdı. Lakin endomiokardial biopsiya SağM divarının prosesə cəlb olunmasındakı müxtəlifliyə görə məhdud həssaslığa malikdir.[27]
1994-cü ildə Beynəlxalq Tövsiyələr ilk dəfə olaraq ARVD üçün ailə anamnezi, aritmiyalar, EKQ anormallıqları, toxuma xüsusiyyətləri, SağM struktur və funksional anormallıqlarına görə böyük və kiçik meyarlar nəzərdə tutmuşdur.[1] Erkənvə ailəvixəstəliyimüəyyənetmək üçünbumeyarlarınspesifikliyiyüksəkolsada, həssaslığı yoxdərəcəsindədir.
2010-cu ildə Rəhbər Tövsiyə meyarlarına normal subyektlərin məlumatları əsasında müəyyən edilmiş kəmiyət meyarları və anormallıqları da daxil olmaqla yenidən baxılmışdır. [28]
Böyük və kiçik meyarlar cədvəl 1-də göstərilmişdir:
ARVD diaqnozu üçün nəzərdə tutulan terminologiyaya 6 müxtəlif kateqoriyadan olan aşağıdakı böyük və kiçik meyarlar aid edilmişdir.
Exokardioqrafiyada, ürək MRT-sində və/yaxud SağM angioqrafiyasında qlobal və ya regional disfunksiya və struktur zədələnmələri (böyük meyar);
Endomiokardial biopsiyada görünən toxumanın xüsusiyyətləri (böyük meyar);
EKQ-də repolyarizasiya anormallıqları (kiçik meyar);
EKQ-də V1-V3 aparmalarında epsilon dalğası yaxud QRS davam müddətinin lokal uzanması (>110msan) (böyük meyar) və yaxud siqnal ortalamalı EKQ-də gec potensiallar (kiçik meyar);
Holter monitorlamada aritmiyalar (kiçik meyar);
Ailə anamnezi. Yaxın qohumlarda autopsiya və ya cərrahiyədə ARVD təsdiq olunmuşsa bu, böyük meyar hesab edilir. SağM displaziyasına şübhəyə görə birinci dərəcəli qohumlarda erkən (<35 yaş) qəfləti ölüm baş verməsi və ikinci dərəcəli qohumlarda hazırkı meyarlara görə ARVD diaqnozu təsdiq olunması ARVD üçün kiçik meyar hesab edilir.
Diaqnoz 2 böyük meyar, 1 böyük və 2 kiçik meyar və ya 4 kiçik meyar olduqda müəyyən olunur.1 böyük və 2 kiçik və ya 3 kiçik meyar olduqda diaqnoz sərhəddi hesab edilir.
Şəkillər

Açar sözlər
İstinadlar
1. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994 Mar. 71(3):215-8. [Medline]. [Full Text].
2. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998 Aug 6. 339(6):364-9. [Medline].
3. Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003 Dec 16. 108(24):3000-5. [Medline].
4. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996 Mar 1. 93(5):841-2. [Medline].
5. Saguner AM, Brunckhorst C, Duru F. Arrhythmogenic ventricular cardiomyopathy: A paradigm shift from right to biventricular disease. World J Cardiol. 2014 Apr 26. 6(4):154-74. [Medline]. [Full Text].
6. Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002 Oct 16. 40(8):1445-50. [Medline].
7. Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007 Apr 3. 115(13):1710-20. [Medline].
8. Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997 Nov 15. 30(6):1512-20. [Medline].
9. Johns Hopkins Medicine. Heart & Vascular Institute. Arrhythmogenic right ventricular dysplasia (arrhythmogenic right ventricular cardiomyopathy). Available at http://www.arvd.com. Accessed: Sept. 11, 2014.
10. Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K, et al. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2006 Oct 3. 48(7):1416-24. [Medline].
11. Moric-Janiszewska E, Markiewicz-Loskot G. Review on the genetics of arrhythmogenic right ventricular dysplasia. Europace. 2007 May. 9(5):259-66. [Medline].
12. Coonar AS, Protonotarios N, Tsatsopoulou A, Needham EW, Houlston RS, Cliff S, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1998 May 26. 97(20):2049-58. [Medline].
13. Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, et al. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J. 1986 Oct. 56(4):321-6. [Medline]. [Full Text].
14. Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010 Feb 9. 55(6):587-97. [Medline]. [Full Text].
15. Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010 Jan. 7(1):22-9. [Medline].
16. Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A, et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006 Jan 24. 113(3):356-64. [Medline].
17. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011 Aug. 13(8):1077-109. [Medline].
18. Thiene G, Basso C, Danieli G, Rampazzo A, Corrado D, Nava A. Arrhythmogenic right ventricular cardiomyopathy a still underrecognized clinic entity. Trends Cardiovasc Med. 1997 Apr. 7(3):84-90. [Medline].
19. Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004 Dec. 97(3):499-501. [Medline].
20. Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005 Dec 20. 112(25):3823-32. [Medline].
21. Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004 Oct 5. 110(14):1879-84. [Medline].
22. Tonet JL, Castro-Miranda R, Iwa T, Poulain F, Frank R, Fontaine GH. Frequency of supraventricular tachyarrhythmias in arrhythmogenic right ventricular dysplasia. Am J Cardiol. 1991 May 15. 67(13):1153. [Medline].
23. Maron BJ, Carney KP, Lever HM, Lewis JF, Barac I, Casey SA, et al. Relationship of race to sudden cardiac death in competitive athletes with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003 Mar 19. 41(6):974-80. [Medline].
24. Muthappan P, Calkins H. Arrhythmogenic right ventricular dysplasia. Prog Cardiovasc Dis. 2008 Jul-Aug. 51(1):31-43. [Medline].
25. Morin DP, Mauer AC, Gear K, Zareba W, Markowitz SM, Marcus FI, et al. Usefulness of precordial T-wave inversion to distinguish arrhythmogenic right ventricular cardiomyopathy from idiopathic ventricular tachycardia arising from the right ventricular outflow tract. Am J Cardiol. 2010 Jun 15. 105(12):1821-4. [Medline].
26. Gerlis LM, Schmidt-Ott SC, Ho SY, Anderson RH. Dysplastic conditions of the right ventricular myocardium: Uhl's anomaly vs arrhythmogenic right ventricular dysplasia. Br Heart J. 1993 Feb. 69(2):142-50. [Medline]. [Full Text].
27. Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J. 1996 Jul. 132(1 Pt 1):203-6. [Medline].
28. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010 Apr 6. 121(13):1533-41. [Medline]. [Full Text].
Məqalə barədə təfərrüatlar:
Nəşr tarixçəsi
Dərc edilib: 31.Dec.2017
Müəllif hüququ
© 2013-2025. Azərbaycan Kardiologiya Cəmiyyətinin rəsmi nəşri. Jurnal "Uptodate in Medicine" tibb nəşriyyatı tərəfindən dərc olunur. Bütün hüquqlar qorunur.Əlaqəli məqalələr
Baxılıb: 1549